
The structures of metal coordination sites
 المؤلف:
Peter Atkins, Tina Overton, Jonathan Rourke, Mark Weller, and Fraser Armstrong
المؤلف:
Peter Atkins, Tina Overton, Jonathan Rourke, Mark Weller, and Fraser Armstrong
 المصدر:
Shriver and Atkins Inorganic Chemistry ,5th E
المصدر:
Shriver and Atkins Inorganic Chemistry ,5th E
 الجزء والصفحة:
ص730-731
الجزء والصفحة:
ص730-731
 2025-10-22
2025-10-22
 64
64
The structures of metal coordination sites
Key point: The likelihood that a protein will coordinate a particular kind of metal centre can be inferred from the amino acid sequence and ultimately from the gene itself. The structures of metal coordination sites have been determined mainly by X-ray diffraction (now mostly by using a synchrotron, Section 8.1) and sometimes by NMR spectros copy.3 The basic structure of the protein can be determined even if the resolution is too low to reveal details of the coordination at the metal site. The packing of amino acids in a protein is far denser than is commonly conveyed by simple representations, as may be seen by comparing the representations of the structure of the K channel in Fig. 27.3. Thus, even the substitution of an amino acid that is far from a metal centre may result in significant structural changes to its coordination shell and immediate environment. Of special interest are channels or clefts that allow a substrate selective access to the active site, pathways for long-range electron transfer (metal centres positioned less than 1.5 nm apart), pathways for long-range proton transfer (comprising chains of basic groups such as carboxylates and water molecules in close proximity, usually less than 0.3 nm apart), and tunnels for small gaseous molecules (which can be revealed by placing the crystal under Xe, an electron-rich gas).
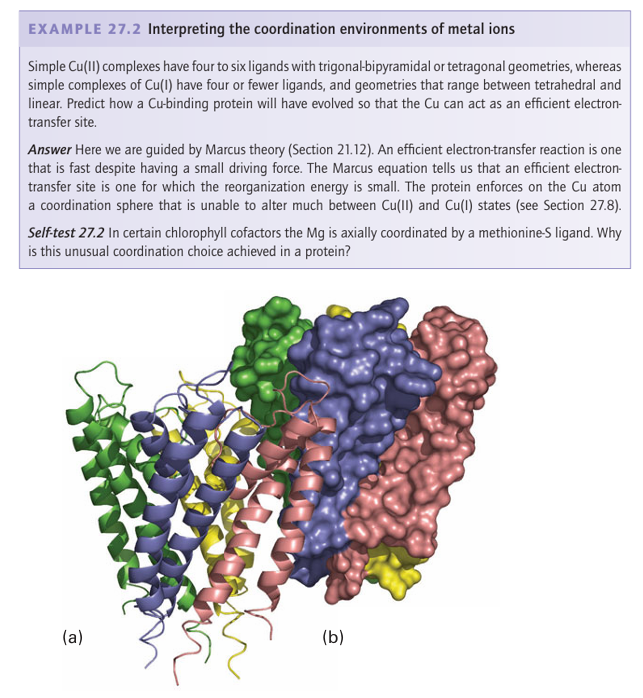
Figure 27.3 Illustrations of how protein structures are represented to reveal either (a) secondary structure or (b) the filling of space by nonhydrogen atoms. The example shows the four subunits of the K channel, which is found mainly embedded in the cell membrane. Other physical methods described in Chapter 8 provide less information on the overall structure but are useful for identifying ligands. Thus, EPR spectroscopy is very import ant for studying d-block metals, especially those engaged in redox chemistry, because at least one oxidation state usually has an unpaired electron. The use of NMR is restricted to proteins smaller than 20–30 kg mol 1 because tumbling rates for larger proteins are too slow and 1H resonances are too broad to observe unless shifted away from the nor mal region (δ=1-10) by a paramagnetic metal centre. Extended X-ray absorption fine structure spectroscopy (EXAFS, Section 8.9) can provide structural information on metal sites in amorphous solid samples, including frozen solutions. Vibrational spectroscopy (Section 8.4) is increasingly being used: IR spectroscopy is particularly useful for ligands such as CO and CN−, and resonance Raman spectroscopy is very helpful when the metal centre has strong electronic transitions, such as occur with Fe porphyrins. Mössbauer spectroscopy (Section 8.7) plays a special role in studies of Fe sites. Perhaps the greatest challenge is presented by Zn2+, which has a d10 configuration that provides no useful magnetic or electronic signatures. Metal ion binding sites can often be predicted from a gene sequence. Bioinformatics, the development and use of software to analyse and compare DNA sequences, is a powerful tool because many proteins that bind metal ions or have a metal-containing cofactor occur at cellular levels below that normally detectable directly by analysis and isolation. A particularly common sequence of the human genome encodes the so-called Zn finger domain, thereby identifying proteins that are involved in DNA binding (Section 27.5). Likewise, it can be predicted whether the protein that is encoded is likely to bind Cu, Ca, an Fe-porphyrin, or different types of Fe S clusters. The gene can be cloned and the protein for which it encodes can be produced in sufficiently large quantities by ‘overex pression’ in suitable hosts, such as the common gut bacterium Escherichia coli or yeast, to enable it to be characterized. Furthermore, the use of genetic engineering to alter the amino acids in a protein, the technique of site-directed mutagenesis, is a powerful principle in biological inorganic chemistry. This technique often permits identification of the ligands to particular metal ions and the participation of other residues essential to functions such as substrate binding or proton transfer. Although structural and spectroscopic studies give a good idea of the basic coordination environment of a metal centre, it is by no means certain that the same structure is retained in key stages of a catalytic cycle, in which unstable states are formed as intermediates. The most stable state of an enzyme, in which form it is usually isolated, is called the ‘resting state’. Many enzymes are catalytically inactive on isolation and must be subjected to an activation procedure that may involve reinsertion of a metal ion or other cofactor or re moval of an inhibitory ligand. Intense efforts have been made to model the active sites of metalloproteins by synthe sizing analogues. The models may be divided into two classes: those designed to mimic the structure and spectroscopic properties of the real site, and those synthesized with the intention of mimicking a functional activity, most obviously catalysis. Synthetic models not only illuminate the chemical principles underlying biological activity but also generate new directions for coordination chemistry. As we shall see throughout this chapter, the difficulty is that an enzyme not only imposes some strain on the coordination sphere of a metal atom (even a porphyrin ring is puckered in most cases) but also provides, at fixed distances, functional groups that provide additional coulombic and hydrogen-bonding interactions essential for binding and activating substrates. Indeed, the active site of a metalloenzyme is arguably the ultimate example of supra molecular chemistry.
 الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة




















