علم الكيمياء

تاريخ الكيمياء والعلماء المشاهير
التحاضير والتجارب الكيميائية
المخاطر والوقاية في الكيمياء
اخرى
مقالات متنوعة في علم الكيمياء
كيمياء عامة
الكيمياء التحليلية
مواضيع عامة في الكيمياء التحليلية
التحليل النوعي والكمي
التحليل الآلي (الطيفي)
طرق الفصل والتنقية
الكيمياء الحياتية
مواضيع عامة في الكيمياء الحياتية
الكاربوهيدرات
الاحماض الامينية والبروتينات
الانزيمات
الدهون
الاحماض النووية
الفيتامينات والمرافقات الانزيمية
الهرمونات
الكيمياء العضوية
مواضيع عامة في الكيمياء العضوية
الهايدروكاربونات
المركبات الوسطية وميكانيكيات التفاعلات العضوية
التشخيص العضوي
تجارب وتفاعلات في الكيمياء العضوية
الكيمياء الفيزيائية
مواضيع عامة في الكيمياء الفيزيائية
الكيمياء الحرارية
حركية التفاعلات الكيميائية
الكيمياء الكهربائية
الكيمياء اللاعضوية
مواضيع عامة في الكيمياء اللاعضوية
الجدول الدوري وخواص العناصر
نظريات التآصر الكيميائي
كيمياء العناصر الانتقالية ومركباتها المعقدة
مواضيع اخرى في الكيمياء
كيمياء النانو
الكيمياء السريرية
الكيمياء الطبية والدوائية
كيمياء الاغذية والنواتج الطبيعية
الكيمياء الجنائية
الكيمياء الصناعية
البترو كيمياويات
الكيمياء الخضراء
كيمياء البيئة
كيمياء البوليمرات
مواضيع عامة في الكيمياء الصناعية
الكيمياء التناسقية
الكيمياء الاشعاعية والنووية

Can We Predict Whether Reactions Will Be Fast or Slow?
 المؤلف:
John D. Roberts and Marjorie C. Caserio
المؤلف:
John D. Roberts and Marjorie C. Caserio
 المصدر:
Basic Principles of Organic Chemistry : LibreTexts project
المصدر:
Basic Principles of Organic Chemistry : LibreTexts project
 الجزء والصفحة:
........
الجزء والصفحة:
........
 23-12-2021
23-12-2021
 2605
2605




+
-
20
Can We Predict Whether Reactions Will Be Fast or Slow?
To a considerable degree, we can predict relative reactivities, provided we use common sense to limit our efforts to reasonable situations. In the preceding section, we argued that reactions in which atoms or radicals combine can well be expected to be extremely fast because each entity has a potentially bonding electron in an outer unfilled shell, and bringing these together to form a bond does not require that other bonds be broken:

Figure 4-6). As CH4 and Cl⋅ get closer together, the new bond starts to form and the old bond starts to break. At the top of the barrier, the hydrogen will be bonded partly to chlorine and partly to carbon, [Cl−−−H−−−CH3], and this we call the activated complex or transition state. The concept of the transition state is an important one, which we will use repeatedly later in connection with many other kinds of reactions. The value of the concept lies in the fact that the reacting system, when it reaches the top of the barrier, can be though of as a chemical entity with a particular, even if not well-defined, structure and definite thermodynamic properties.
The difference between the average energy of the reactants and the energy of the transition state is called the activation energy (Figure 4-4). We expect this energy to be smaller (lower barrier) if a weak bond is being broken and a strong bond is being made. The perceptive reader will notice that we are suggesting a parallel between reaction rate and ΔH0 because ΔH0 depends on the difference in strengths of the bonds being broken and formed. Yet previously , we pointed out that the energy barrier for a reaction need bear no relationship to how energetically feasible the reaction is, and this is indeed true for complex reactions involving many steps. But our intuitive parallel between rate and ΔH0ΔH0 usually works quite well for the rates of individual steps. This is borne out by experimental data on rates of removal of a hydrogen atom from methane by atoms or radicals (X⋅), such as F⋅, Cl⋅, Br⋅, HO⋅, H2N⋅, which generally parallel the strength of the new bond formed:
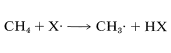
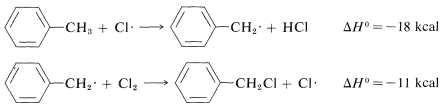
Similarly, if we look at the H−C bond-dissociation energies of the hydrocarbons shown in Table 4-6, we would infer that Cl⋅ would remove a hydrogen most rapidly from the carbon forming the weakest C−H bond and, again, this is very much in accord with experience. For example, the chlorination of methylbenzene (toluene) in sunlight leads to the substitution of a methyl hydrogen rather than a ring hydrogen for the reason that the methyl C−H bonds are weaker and are attacked more rapidly than the ring C−H bonds. This can be seen explicitly in the ΔH0 values for the chain-propagation steps calculated from the bond-dissociation energies of Table 4-6.
Methyl substitution (observed):
Ring substitution (not observed):
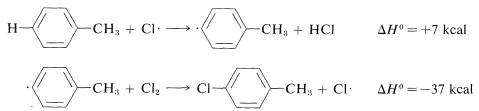
The ΔH0 of ring-hydrogen abstraction is unfavorable by +7kcal because of the high C−H bond energy (110kcal). Thus this step is not observed. It is too slow in comparison with the more favorable reaction at the methyl group even though the second propagation step is energetically favorable by −37kcal and presumably would occur very rapidly. Use of bond-dissociation energies to predict relative reaction rates becomes much less valid when we try to compare different kinds of reactions. To illustrate, ethane might react with F⋅ to give fluoromethane or hydrogen fluoride:
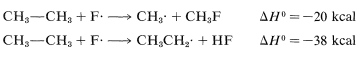
It is not a good idea to try to predict the relative rates of these two reactions on the basis of their overall ΔH0 values because the nature of the bonds made and broken is too different.
 الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)